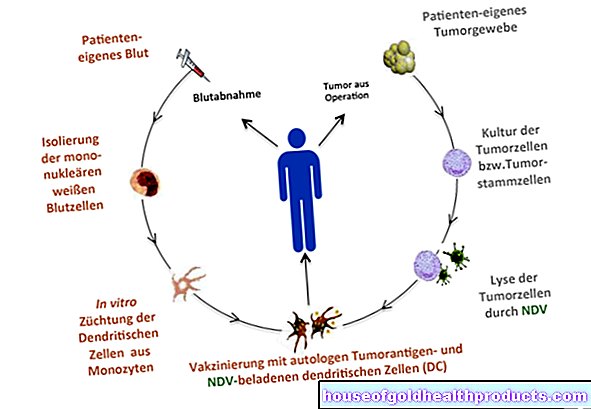
Asuntos Regulatorios
Martina Feichter estudió biología con una asignatura optativa de farmacia en Innsbruck y también se sumergió en el mundo de las plantas medicinales. De ahí no fue lejos para otros temas médicos que aún la cautivan hasta el día de hoy. Se formó como periodista en la Academia Axel Springer en Hamburgo y ha estado trabajando para desde 2007, primero como editora y desde 2012 como escritora independiente.
Más sobre los expertos de Todo el contenido de es verificado por periodistas médicos.
La búsqueda de nuevos principios activos frente a determinadas enfermedades o dolencias es tediosa y no siempre acaba con éxito. De 5,000 a 10,000 candidatos que se prueban en los laboratorios de investigación de las compañías farmacéuticas, en promedio solo uno termina como medicamento terminado en la farmacia. Y hay un promedio de 13,5 años en el medio.

Busque el "objetivo"
Incluso antes de que se realicen pruebas con nuevas sustancias, los investigadores piensan en las propiedades de la sustancia en cuestión y qué reacción debería desencadenar en el organismo. Esto puede ser, por ejemplo, reducir la presión arterial, bloquear una determinada sustancia mensajera o liberar una hormona.
Con este fin, los investigadores están buscando un "objetivo" adecuado, es decir, un punto de ataque en el proceso de la enfermedad, en el que un ingrediente activo pueda adherirse y así influir positivamente en el proceso de la enfermedad. En la mayoría de los casos, el objetivo es una enzima o un receptor (punto de acoplamiento en las células para las hormonas u otras sustancias mensajeras). A veces, al paciente también le falta una determinada sustancia. Luego, rápidamente queda claro que el medicamento que está buscando debe compensar esta deficiencia. Un ejemplo bien conocido es la insulina en personas con diabetes (diabetes mellitus).
Busque el ingrediente activo
Tan pronto como se ha determinado un objetivo, los científicos buscan un ingrediente activo que pueda actuar en el punto de ataque elegido (detección). Eso generalmente significa: prueba, prueba, prueba. Cada día se examinan hasta 300.000 sustancias diferentes para comprobar su idoneidad (cribado de alto rendimiento = HTS). De estas, aproximadamente cada sustancia 200 a 1000 muestra un efecto en el objetivo seleccionado, aunque a veces solo sea muy pequeño. Este tipo de acierto se denomina "acierto".
Las sustancias de ensayo se producen principalmente de forma química, es decir, sintética. Desde hace algún tiempo, las sustancias modificadas genéticamente también han ido ganando importancia. Se obtienen con la ayuda de células modificadas genéticamente (como ciertas bacterias) y forman la base de los biofarmacéuticos (fármacos biológicos).
mejoramiento
En la mayoría de los casos, los "hits" encontrados aún deben optimizarse. A veces, por ejemplo, la eficacia de una sustancia puede aumentar si su estructura se modifica ligeramente. En estos experimentos, los científicos a menudo trabajan con simulaciones por computadora, con la ayuda de las cuales se puede estimar de antemano el efecto de un cambio químico en la sustancia. Si el pronóstico es bueno, la sustancia se ajusta en la vida real, es decir, en el laboratorio. A continuación, se vuelve a examinar su efecto sobre el objetivo.
De esta forma, los investigadores mejoran gradualmente una nueva sustancia activa, lo que suele llevar varios años.En el mejor de los casos, eventualmente llegarán al punto en el que la sustancia está lista para el siguiente paso: se presenta una solicitud de patente y luego se somete a estudios preclínicos como un llamado candidato a ingrediente activo.
Estudios preclínicos
En la fase de desarrollo preclínico (preclínico), el fármaco candidato se prueba en tubos de ensayo (por ejemplo, en cultivos celulares) y en animales. Por un lado, se trata de cuestiones farmacológicas, por ejemplo, qué le sucede a la sustancia en las células o en todo un organismo:
- ¿Cómo se recibe?
- ¿Cómo se distribuye en el organismo?
- ¿Qué reacciones desencadena?
- ¿Será modificado o desmantelado?
- ¿Será eliminado?
Por otro lado, los científicos están investigando exactamente qué efecto tiene la sustancia en el objetivo, cuánto dura y qué dosis es necesaria para ello.
Sin embargo, sobre todo, los estudios preclínicos sirven para responder preguntas sobre la toxicidad (toxicidad) del fármaco candidato. ¿Es venenosa la sustancia? ¿Puede causar cáncer? ¿Puede cambiar genes? ¿Puede dañar un embrión o un feto?
Muchos candidatos a fármacos no pasan las pruebas de toxicidad. Solo las sustancias que pasan todas las pruebas de seguridad pueden ingresar a la siguiente fase de desarrollo con estudios en humanos (estudios clínicos).
Siempre que sea posible, las pruebas preclínicas se realizan en tubos de ensayo, por ejemplo en cultivos celulares, fragmentos de células u órganos humanos aislados. Sin embargo, algunas preguntas solo se pueden responder en pruebas en un organismo vivo, y los experimentos con animales son necesarios para esto.
Estudios clínicos
El fármaco candidato se está probando en humanos por primera vez en estudios clínicos. Se hace una distinción entre tres fases de estudio que se complementan entre sí:
- Fase I: el fármaco candidato se prueba en unos pocos voluntarios sanos (sujetos de prueba).
- Fase II: a esto le siguen pruebas en algunas personas enfermas (por ejemplo, en pacientes con presión arterial alta si el candidato a fármaco se convertirá en un nuevo agente antihipertensivo).
- Fase III: Ahora la prueba se realiza en un gran número de personas enfermas.
Cada fase del estudio debe ser aprobada previamente por los organismos responsables: Por un lado, esto incluye a la autoridad nacional responsable, dependiendo del fármaco candidato, ya sea el Instituto Federal de Medicamentos y Productos Sanitarios (BfArM) o el Instituto Paul Ehrlich (PEI). ). Por otro lado, todo estudio clínico necesita el permiso de un comité de ética (formado por médicos, abogados, teólogos y laicos). Este procedimiento está destinado a proteger a los participantes del estudio de la mejor manera posible.
El fabricante farmacéutico que desarrolló el fármaco candidato puede realizar los estudios clínicos por sí mismo. O contrata a una "Organización de Investigación Clínica" (CRO) para hacer esto. Esta es una empresa que se especializa en la realización de estudios clínicos.
Estudios de fase I
Por lo general, entre 60 y 80 adultos sanos que se han ofrecido como voluntarios para este acto actúan como personas de prueba en la fase I. Después de una explicación completa y el consentimiento de los participantes del estudio, inicialmente solo se les da una pequeña cantidad de ingrediente activo.
En hasta 30 pruebas consecutivas, los científicos verifican si los hallazgos de las pruebas en el tubo de ensayo y en animales también se pueden transferir a los humanos, es decir, si el ingrediente activo se absorbe, distribuye, convierte y excreta nuevamente como lo haría en el estudio preclínico. Pruebas determinadas. Además, se investiga qué tan bien toleran las personas de prueba el candidato a fármaco.
¿Tableta, jeringa o ungüento?
Después de completar con éxito la fase I, entran en juego los llamados galénicos: los científicos ahora están trabajando en el "empaquetado" óptimo para el ingrediente activo: ¿debe administrarse por vía intravenosa en forma de tableta, cápsula, supositorio, jeringa o infusión?
La respuesta a esta pregunta es muy importante: la forma de dosificación tiene una gran influencia en la confiabilidad, la rapidez y la duración de la función del ingrediente activo en el cuerpo. También afecta el tipo y la gravedad de los posibles efectos secundarios. Algunos ingredientes activos se toleran mucho mejor como inyección que cuando ingresan al cuerpo en forma de tabletas a través del tracto gastrointestinal.
Además, los expertos galénicos comprueban si deben añadirse sustancias auxiliares a la nueva preparación y qué sustancias auxiliares. Por ejemplo, algo que mejore el sabor del fármaco o actúe como portador o conservante.
Puede leer más sobre la búsqueda del "envase" adecuado para un nuevo ingrediente activo y para materiales auxiliares adecuados en el artículo Galénica - Fabricación de productos farmacéuticos.
Estudios de fase II y fase III
Después de los sujetos sanos en la fase I, es el turno de los enfermos de la fase II para probar el candidato a fármaco:
- Fase II: aquí, el nuevo fármaco candidato se prueba en la mayoría de los casos de 100 a 500 pacientes. La atención se centra en la eficacia, la dosis óptima y la tolerancia de la preparación.
- Fase III: Aquí se llevan a cabo los mismos controles que en la Fase II, solo que en un número significativamente mayor de pacientes (varios miles). Además, se presta atención a las posibles interacciones con otros fármacos.
En ambas fases, los diferentes tratamientos se comparan entre sí: solo algunos de los pacientes reciben la nueva preparación, el resto recibe una medicación estándar habitual o familiar o un placebo, una preparación que se ve exactamente como la nueva pero no contiene ninguna ingrediente activo (fármaco ficticio). Como regla general, ni el paciente ni el médico tratante saben quién recibe qué. Estos "estudios doble ciego" están diseñados para evitar que las esperanzas, los temores o las actitudes escépticas de los médicos y pacientes influyan en el resultado del tratamiento.
Concesión de aprobación
Incluso si un nuevo medicamento ha pasado todos los estudios y pruebas requeridos, no se puede vender simplemente. Para ello, la empresa farmacéutica debe solicitar primero la aprobación del medicamento a la autoridad competente (consulte a continuación: Opciones de aprobación). Esto verifica cuidadosamente todos los resultados del estudio y, en el mejor de los casos, otorga permiso al fabricante para llevar el nuevo medicamento al mercado.
Fase IV
Incluso después de que se haya aprobado un medicamento, las autoridades y la compañía farmacéutica vigilan la nueva preparación, por ejemplo, con respecto a los efectos secundarios raros. Estos son efectos indeseables que ocurren en menos de 1 de cada 10,000 pacientes tratados y, por lo tanto, son apenas detectables en las fases previas del estudio (con grupos de pacientes más pequeños). Los médicos deben informar cualquier efecto secundario imprevisto de un medicamento.
Si es necesario, la autoridad de homologación le pedirá al fabricante que indique estos efectos secundarios recién descubiertos en el prospecto. Sin embargo, también puede imponer restricciones de uso: si, por ejemplo, se descubren efectos secundarios raros pero graves en el área del riñón, las autoridades pueden ordenar que el medicamento ya no se use en personas con enfermedades renales existentes.
En casos extremos, las autoridades pueden retirar la aprobación de un medicamento por completo si, con el tiempo, han surgido riesgos inaceptables de su uso. A veces, el fabricante retira voluntariamente dicho producto del mercado.
Los médicos también usan registros para registrar cómo les está yendo al nuevo medicamento en la vida diaria de sus pacientes. El fabricante utiliza los resultados de dichos estudios observacionales, por ejemplo, para mejorar la dosificación o la forma de dosificación de la preparación.
A veces, la práctica diaria también muestra que el ingrediente activo ayuda contra otras enfermedades. El fabricante generalmente continúa investigando en esta dirección, con nuevos estudios de fase II y III. Si tiene éxito, también puede solicitar la aprobación de esta nueva indicación.
Opciones de aprobación
En principio, una empresa farmacéutica puede solicitar la aprobación de un nuevo medicamento para toda la UE o solo para un solo estado miembro:
Proceso de aprobación centralizado
La aprobación del medicamento se solicita aquí directamente a la Agencia Europea de Medicamentos (EMA). Las autoridades de aprobación de los estados miembros de la UE también participan en la prueba posterior. Si se aprueba la aplicación, la preparación se puede vender en cualquier lugar de la UE. Este proceso de aprobación lleva una media de un año y medio y es obligatorio para algunos medicamentos (por ejemplo, para los preparados fabricados biotecnológicamente y para los medicamentos contra el cáncer con nuevos ingredientes activos).
Proceso de aprobación nacional
La solicitud de aprobación se envía a las autoridades nacionales y, por lo tanto, solo en el país en cuestión. En Alemania, el Instituto Federal de Medicamentos y Productos Sanitarios (BfArM) y el Instituto Paul Ehrlich (PEI) son responsables de esto. El BfArM se ocupa de la mayoría de los productos farmacéuticos humanos, el PEI se encarga de los sueros, vacunas, alérgenos de prueba, sueros de prueba y antígenos de prueba, sangre y productos sanguíneos, tejidos y medicamentos para terapia génica y terapia celular.
Aprobación de medicamentos en varios países de la UE
Además, existen otras dos opciones si una empresa farmacéutica desea obtener la aprobación en varios países de la UE:
- Procedimiento descentralizado: en un "procedimiento descentralizado" (DCP), una empresa farmacéutica puede solicitar la aprobación nacional de un nuevo medicamento en varios países del Espacio Económico Europeo al mismo tiempo.
- Procedimiento de Reconocimiento Mutuo: Si un medicamento ya tiene una aprobación nacional en un país del Espacio Económico Europeo, esto puede ser reconocido por otros estados miembros en el marco del "Procedimiento de Reconocimiento Mutuo" (MRP).
La solicitud de aprobación de un nuevo fármaco es muy cara para las empresas farmacéuticas. Por ejemplo, procesar una solicitud de aprobación de un ingrediente activo completamente nuevo en la EMA cuesta alrededor de 260.000 euros en el caso más simple.
Aprobación estándar
Algunos medicamentos se lanzan a la venta mediante una aprobación estándar: no se trata de preparaciones desarrolladas recientemente, sino de aquellas cuya fabricación se basa en determinadas monografías estipuladas por el legislador. Además, estos medicamentos no deben representar ningún peligro para los seres humanos o los animales. En una monografía (por ejemplo, para supositorios de paracetamol 250 mg), entre otras cosas, la composición y la dosis de la preparación en cuestión se definen con precisión, al igual que el área de aplicación.
Si se cumplen todos estos requisitos, el fabricante no tiene que solicitar su propia aprobación individual de medicamentos. Esto le permite llevar medicamentos al mercado a un precio muy asequible. Existen aprobaciones estándar para tabletas de carbón vegetal (250 mg), gotas para los ojos y soluciones de atropina en varias concentraciones, así como supositorios de paracetamol y tabletas de ácido acetilsalicílico en varias dosis.
Los farmacéuticos, por ejemplo, también pueden preparar una solución salina de acuerdo con las instrucciones de la farmacopea correspondiente y luego venderla. Sin embargo, debe indicar el uso de dicha aprobación estándar a la autoridad de aprobación y la autoridad estatal responsable.
Otras formas de obtener aprobaciones de medicamentos
En la UE, además del procedimiento de aprobación convencional, también existen opciones para hacer que un nuevo medicamento esté disponible antes de lo habitual. Estas no son solo aprobaciones rápidas. Por el contrario, se están haciendo intentos de diversas formas para garantizar que los afectados puedan beneficiarse de los ingredientes activos incluso sin la aprobación de un fármaco tradicional. Los expertos hablan de las llamadas vías adaptativas:
Programas de uso compasivo
Aquí, pacientes muy específicos reciben medicamentos que en realidad aún se encuentran en ensayos clínicos. El requisito previo es que ya no exista ninguna otra opción de tratamiento y el paciente no pueda participar en un estudio correspondiente sobre este fármaco. Estas exenciones deben solicitarse por separado para cada paciente individual.
Aprobación condicional de medicamentos
Esto es, por así decirlo, una rápida aprobación. Las estrictas pruebas de eficacia y seguridad no tienen por qué estar presentes en la medida en que de otro modo es habitual. Sin embargo, se aplican ciertas condiciones:
- La aprobación condicional de medicamentos está limitada en el tiempo.
- El fabricante debe proporcionar los documentos faltantes que son necesarios para la aprobación regular de medicamentos.
La aprobación condicional se utiliza, por ejemplo, en pandemias para proporcionar rápidamente un medicamento adecuado contra la enfermedad infecciosa.
Aprobación en circunstancias excepcionales
Esta ruta especial está disponible, por ejemplo, para enfermedades raras. Dado que hay muy pocas personas enfermas, la compañía farmacéutica no puede enviar la cantidad de datos que de otro modo sería necesaria para su examen. Sin embargo, con la aprobación de este medicamento, el fabricante generalmente tiene que verificar anualmente si hay nuevos datos y hallazgos.
Aprobación acelerada de medicamentos (evaluación acelerada)
El comité responsable de la EMA verifica y evalúa los documentos de aprobación más rápidamente, en lugar de los 210 habituales en 150 días. Este camino es posible si existe un ingrediente activo prometedor contra una enfermedad que hasta ahora no podría tratarse adecuadamente.
Medicamentos prioritarios (PRIME)
En los casos en los que aún no se ha satisfecho una necesidad, la EMA y el fabricante de medicamentos pueden trabajar juntos desde el principio, incluso durante las primeras pruebas. De esta manera, los expertos pueden evaluar la eficacia y la seguridad en una etapa temprana e iniciar más procedimientos más rápidamente si el fármaco demuestra ser prometedor.
Revisión continua (revisión continua)
En el caso de medicamentos y vacunas que se necesitan con urgencia, la EMA puede, como ya se señaló, aprobar "condicionalmente" los ingredientes activos o trabajar con los fabricantes en una etapa temprana antes de la aprobación final. En casos importantes, el llamado proceso de revisión continua comienza antes de estas aprobaciones. Los expertos evalúan los datos existentes antes de que el fabricante pueda enviar todos los documentos relevantes para su aprobación. Además, verifican continuamente todos los nuevos resultados obtenidos de estudios posteriores.
Por ejemplo, la EMA aplicó el proceso de revisión continua a la aprobación condicional del medicamento viral remdesivir durante la pandemia de coronavirus. Como parte del proceso de aprobación de las vacunas corona, los expertos también verificaron los resultados que ya estaban disponibles y luego se obtuvieron durante los estudios de fase III en curso.
Medicamentos para niños
Los medicamentos nuevos generalmente pasan por varios estudios antes de que se les permita salir al mercado. Sin embargo, durante mucho tiempo, un grupo de pacientes recibió menos atención en la investigación: niños y adolescentes. Para el tratamiento de menores, la dosis de un medicamento que se ha probado en adultos a menudo simplemente se redujo.
Sin embargo, desde 2007, cada nuevo fármaco en la UE ha tenido que probarse en menores en estudios de fase II y III si se va a utilizar más tarde en este grupo de edad. Las pruebas en niños o adolescentes a menudo solo se inician una vez que se han completado con éxito los estudios de fase II en adultos. Un grupo separado de expertos de la Agencia Europea de Medicamentos EMA, el Comité Pediátrico, decide los detalles.
Las pruebas de admisión en menores tienen sentido porque los cuerpos de niños y adolescentes a menudo reaccionan de manera diferente a una droga que el de los adultos. Por tanto, la eficacia y la tolerabilidad pueden ser diferentes. Por lo tanto, la dosis debe ajustarse normalmente para los menores. En muchos casos, se requiere una forma diferente de administración para los medicamentos para niños, como gotas o un polvo en lugar de las tabletas grandes que reciben los pacientes adultos.
Hierbas medicinales
Al desarrollar nuevos medicamentos a base de plantas (agentes fitoterapéuticos), la prueba de eficacia, tal como se prescribe en forma de estudios clínicos, es difícil:
Si bien los medicamentos químicos generalmente no contienen más de una o dos sustancias puras, cada planta produce una mezcla de sustancias activas. La mayoría de las veces, esta mezcla también varía en diferentes partes de la planta. Por ejemplo, la hierba de ortiga puede afectar los riñones, mientras que la raíz de ortiga puede afectar el metabolismo hormonal de la próstata. Además, estas mezclas de principios activos varían mucho en función del origen y preparación de la planta, lo que también influye en la eficacia.
En 1978 se creó un grupo de expertos, la denominada Comisión E, para aclarar estas cuestiones. Estos contienen la información conocida en ese momento sobre la composición, efectos y posibles efectos secundarios de las distintas plantas medicinales.
Dado que las monografías de la Comisión E no se han actualizado desde 1994, se utilizan en su lugar las monografías del "Comité de Medicamentos a Base de Hierbas" (HMPC). Este es el comité de la Agencia Europea de Medicamentos responsable de los medicamentos a base de plantas. Se encarga de la evaluación científica de dichos fármacos.
Debe hacerse una distinción entre los medicamentos tradicionales a base de plantas y los medicamentos modernos a base de plantas: en lugar de la aprobación, se requiere el registro. Más sobre esto en la siguiente sección.
Registro en lugar de admisión
Los medicamentos tradicionales a base de plantas y los preparados homeopáticos están exentos del requisito de autorización como productos medicinales de "terapias especiales". En su lugar, debe registrarse:
Para ello, al igual que para la aprobación de medicamentos "normales", se debe presentar una prueba de la inocuidad y la calidad farmacéutica adecuada del medicamento homeopático o tradicional a base de plantas.
En el caso de los medicamentos tradicionales a base de plantas, el efecto farmacológico o la eficacia también debe demostrarse de manera plausible, utilizando lo que se conoce como evidencia tradicional. Esto significa que el fabricante debe utilizar información bibliográfica para demostrar, entre otras cosas, que el medicamento tradicional a base de plantas se ha utilizado médicamente en la UE durante al menos 30 años, incluidos al menos 15 años.
Sin embargo, los estudios clínicos para demostrar la efectividad, según lo prescrito por la aprobación clásica de medicamentos, no son necesarios para los medicamentos homeopáticos ni para los medicamentos tradicionales a base de hierbas para que una empresa pueda venderlos.
A diferencia de los medicamentos tradicionales en la medicina convencional, los remedios alternativos generalmente carecen de evidencia científica extensa de su efectividad, especialmente porque no se requiere un proceso de aprobación de medicamentos que requiera mucho tiempo.
Etiquetas: sintomas valores de laboratorio fitness deportivo