Enfermedad de Hirschsprung
Astrid Leitner estudió medicina veterinaria en Viena. Después de diez años en la práctica veterinaria y el nacimiento de su hija, se pasó, más por casualidad, al periodismo médico. Rápidamente quedó claro que su interés por los temas médicos y su amor por la escritura eran la combinación perfecta para ella. Astrid Leitner vive con una hija, un perro y un gato en Viena y Alta Austria.
Más sobre los expertos de Todo el contenido de es verificado por periodistas médicos.La enfermedad de Hirschsprung es una enfermedad congénita en la que faltan células nerviosas en la última sección del colon. Las personas afectadas a menudo sufren de estreñimiento permanente hasta obstrucción intestinal desde el nacimiento. En la mayoría de los casos, la cirugía es fundamental. ¡Lea todo lo que necesita saber sobre el pronóstico, los síntomas y el tratamiento del "megacolon congénito" aquí!
Códigos ICD para esta enfermedad: los códigos ICD son códigos reconocidos internacionalmente para diagnósticos médicos. Se pueden encontrar, por ejemplo, en cartas médicas o en certificados de incapacidad laboral. Q43

Breve descripción
- ¿Qué es la enfermedad de Hirschsprung?: Malformación congénita de la sección más baja del intestino grueso
- Pronóstico: con un tratamiento oportuno y controles regulares, el pronóstico es bueno.
- Síntomas: náuseas, vómitos, distensión abdominal, evacuación retardada o ausente de las primeras heces del recién nacido ("Kindspech"), estreñimiento, obstrucción intestinal, dolor abdominal
- Causas: falta de células nerviosas en la última sección del colon.
- Factores de riesgo: trisomía 21 (u otros defectos cromosómicos raros), predisposición genética
- Diagnóstico: síntomas típicos, examen físico, radiografías, ultrasonido, medición de la presión en el recto, biopsia, examen genético
- Tratamiento: En la mayoría de los casos, es necesaria una cirugía.
- Prevención: la enfermedad de Hirschsprung es una enfermedad congénita, la prevención no es posible.
¿Qué es la enfermedad de Hirschsprung?
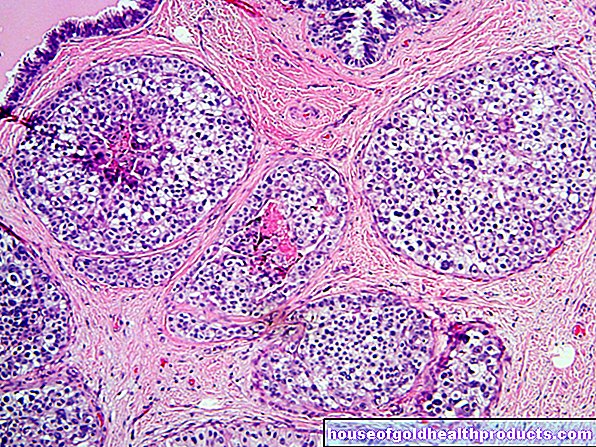
La enfermedad de Hirschsprung (HM), también conocida como megacolon congénito, es una malformación congénita que afecta la última sección del intestino grueso y el esfínter interno del ano. En estos puntos faltan las células nerviosas que normalmente controlan los músculos intestinales. Los músculos intestinales aseguran que el contenido intestinal se transporte más hacia el recto. Para hacer esto, los músculos se contraen en forma de onda y, por lo tanto, empujan más el contenido intestinal. Los médicos llaman a este proceso peristalsis.
En los pacientes con HM, la peristalsis se altera debido a la falta de células nerviosas. El lugar donde faltan los nervios se estrecha permanentemente (estenosis funcional). Dependiendo del tamaño de la sección del intestino afectada, el contenido del intestino no se transporta más allá o solo en cantidades muy pequeñas.
Las heces se acumulan cada vez más frente al punto estrechado y expanden la sección de intestino frente a él. Esta gran expansión del intestino grueso también se conoce como megacolon. Dado que es una enfermedad congénita, los médicos también hablan del "megacolon congénito" o "megacolon congénito". El nombre de enfermedad de Hirschsprung se remonta al pediatra Harald Hirschsprung, quien describió la enfermedad por primera vez en 1888.
Si el intestino no se vacía por completo, se acumulan más y más heces. En la sección estirada y llena de heces del intestino, se puede desarrollar inflamación (enterocolitis), en el peor de los casos una obstrucción intestinal. Si no se trata, esto en última instancia conduce a una intoxicación de la sangre (sepsis) oa un sobrecrecimiento bacteriano del intestino grueso que pone en peligro la vida (megacolon tóxico).
La duración de la sección del intestino afectada en la enfermedad de Hirschsprung varía de un paciente a otro. Sin embargo, es típico que la falta de células nerviosas siempre comience en el ano y se extienda hacia arriba en diferentes grados en el intestino. Dado que el ano también se ve afectado, el músculo del esfínter interno carece de la capacidad de relajarse, lo que dificulta aún más la defecación. La porción de intestino que no funciona también se conoce como segmento sin células nerviosas (agangliónico), razón por la cual los médicos también hablan de aganglionosis.
frecuencia
La enfermedad de Hirschsprung es una de las enfermedades raras. Aproximadamente uno de cada 5.000 niños nace con ella cada año. Los niños tienen cuatro veces más probabilidades de verse afectados que las niñas.
Texto de inicio (avance): La enfermedad de Hirschsprung es una enfermedad congénita en la que faltan células nerviosas en la última sección del colon. Las personas afectadas a menudo sufren de estreñimiento permanente hasta obstrucción intestinal desde el nacimiento. En la mayoría de los casos, la cirugía es fundamental. ¡Lea todo lo que necesita saber sobre el pronóstico, los síntomas y el tratamiento del "megacolon congénito" aquí!
¿Cuál es la esperanza de vida de los pacientes con HM?
Si la enfermedad de Hirschsprung se trata a tiempo, el pronóstico es bueno y la esperanza de vida no está restringida. Sin embargo, la enfermedad requiere atención de seguimiento a largo plazo y controles periódicos de por vida, incluso después de una operación exitosa. En la mayoría de los casos, la cirugía puede restaurar completamente la función intestinal.
Síntomas
Los síntomas que se presentan dependen del tamaño de la sección afectada del intestino y de la cantidad de células nerviosas que faltan. En la mayoría de los casos, los síntomas típicos aparecen en los primeros días de vida.
Los síntomas típicos en los bebés son:
El meconio ("Kindspech") es la primera materia fecal de un recién nacido. No es una evacuación intestinal clásica, sino una acumulación de células de la piel y de las membranas mucosas, jugos digestivos y líquido amniótico ingerido. El meconio es típicamente de color negro verdoso, inodoro y se excreta en los dos primeros días de vida.
Si el meconio no desaparece después del segundo día, puede ser enfermedad de Hirschsprung. En los recién nacidos, un abdomen notablemente grueso y distendido también puede indicar HM.
Los síntomas típicos en los niños son:
Si solo se ve afectada una pequeña sección del intestino, es posible que los padres no noten la malformación de inmediato. Los cambios en las deposiciones a menudo solo se notan después del período de lactancia. La razón: mientras los niños sean amamantados, sus heces son relativamente delgadas y, por lo tanto, más fáciles de transportar. A veces, una constricción en el intestino no se nota. Los problemas solo surgen cuando se cambia la comida. Los niños afectados pueden desarrollar los siguientes síntomas y afecciones:
- Heces duras o pastosas, pastosas (pastosas), ocasionalmente también líquidas
- Estreñimiento constante, evacuaciones intestinales solo cada dos o tres días
- La evacuación de las heces solo es posible con ayudas (enema, inserción de dedos o termómetro)
- Las heces huelen muy mal
- dolor de estómago
- Vómito
- Negativa a comer
- Mal estado general
Causas y factores de riesgo
La causa de la enfermedad de Hirschsprung es que faltan células nerviosas en el intestino grueso. Los contenidos intestinales apenas o ya no se transportan a las zonas afectadas y se acumulan. Dependiendo de cuántas células nerviosas falten, se ven afectadas secciones del intestino grueso de diferentes longitudes.
causas
La razón de la falta de células nerviosas es un desarrollo embrionario alterado entre la cuarta y la duodécima semana de embarazo. No está del todo claro cómo ocurre esto. Los médicos asumen que varios factores influyen:
- Causas genéticas: la enfermedad de Hirschsprung es una enfermedad que se presenta en familias. El riesgo de tener un hijo con HM después de tener otro hijo con la misma enfermedad es del cuatro por ciento. Básicamente, cuanto más largo sea el intestino sin células nerviosas, mayor será el riesgo de transmitir la malformación a su descendencia.
- Reducción del flujo sanguíneo al embrión.
- Infecciones virales en el útero.
- Trastorno de la maduración
Factores de riesgo
La mayoría de las veces, la enfermedad de Hirschsprung se presenta como una enfermedad independiente. Los niños que nacen con trisomía 21 ("síndrome de Down"), sin embargo, tienen un mayor riesgo de enfermedad de Hirschsprung: aproximadamente un tercio de los pacientes con HM están asociados con trisomía 21 u otras enfermedades genéticas como el síndrome de Undine, el síndrome de Shah Waardenburg o el síndrome de Mowat Wilson. .
diagnóstico
En la mayoría de los casos, los primeros síntomas aparecen en la infancia. El pediatra es el primer punto de contacto si se sospecha HM. La historia clínica y los síntomas típicos dan los primeros indicios de la enfermedad de Hirschsprung. El pediatra confirma el diagnóstico con exámenes adicionales:
Examen físico: el médico primero palpa el estómago y presta atención a los cambios típicos, como hinchazón del estómago o bolas de heces duras palpables. Luego realiza un examen "digito-rectal": examina el ano y el recto con el dedo. Un recto estrecho que no contiene heces puede ser un signo de la enfermedad de Hirschsprung. Los ruidos intestinales claramente audibles también indican la enfermedad intestinal.
Examen de ultrasonido: un examen del abdomen mediante ultrasonido le permite al médico detectar un ensanchamiento o un llenado fuerte del intestino con heces. También puede ver si el intestino se está moviendo (peristaltismo intestinal).
Examen de rayos X del intestino grueso (rayos X del colon): para ver si el intestino se ha estrechado en un área determinada, el médico inyecta un medio de contraste en el intestino a través de un tubo delgado. Si se encuentran constricciones en forma de embudo en la radiografía, esto es una indicación de la enfermedad de Hirschsprung.
Extracción de tejido de la pared intestinal (biopsia): A partir de la sexta semana de vida, es posible una colonoscopia con extracción simultánea de tejido (biopsia). Para hacer esto, el médico toma una muestra de tejido de la pared intestinal y usa un microscopio y pruebas especiales para examinar si hay células nerviosas presentes. El diagnóstico se realiza cuando no hay (o hay muy pocas) células nerviosas en la pared intestinal.
Medición de la presión rectal (manometría rectal): durante este examen, el médico verifica si hay peristaltismo y si los dos esfínteres del ano están funcionando. En el caso de la HM, la pared intestinal se contrae como un espasmo, los músculos del esfínter están fuertemente cerrados. Importante: la manometría rectal tiene una importancia limitada para bebés y niños, ya que este método de examen requiere la cooperación del paciente.
Diagnóstico genético molecular: aproximadamente un tercio de los pacientes con HM se ven afectados simultáneamente por la trisomía 21 (u otros defectos cromosómicos raros). Estos se pueden determinar mediante métodos genéticos moleculares (análisis de sangre), pero no los cambios genéticos que desencadenan la HM. Hasta el día de hoy, estos aún no se han explorado completamente.
tratamiento
En la mayoría de los casos de enfermedad de Hirschsprung, es necesaria la cirugía. El momento en que se realiza el procedimiento depende principalmente del estado general del paciente.
Cirugía inmediata si el estado general es bueno
Los pacientes con HM suelen ser operados pronto para que no haya complicaciones como una obstrucción intestinal. Durante la operación, el cirujano extirpa la sección afectada del intestino y, por lo tanto, también la constricción, de modo que las evacuaciones intestinales normales sean posibles nuevamente. El cirujano realiza el procedimiento a través del ano o de la cavidad abdominal.
Medidas puente hasta la operación si el estado general es malo
Si el estado general es malo (recién nacido débil) o si ya han ocurrido complicaciones como envenenamiento de la sangre (sepsis), puede ser necesario esperar un tiempo antes de la operación. En estos casos, el médico extrae las heces con regularidad hasta la operación enjuagando el intestino (enema) o ensanchando la sección intestinal con instrumentos especiales.
A veces es necesario crear temporalmente un ano artificial (estoma) antes de la operación real.
Después de la operación
Se necesitan algunas semanas para que la herida quirúrgica en el intestino sane y para que el intestino grueso vuelva a funcionar normalmente. Durante este tiempo, los padres de los niños afectados deben observar los siguientes puntos:
- Evacuaciones intestinales frecuentes: muchos niños tienen evacuaciones intestinales muy frecuentes (de cinco a 30 veces) y delgadas después de la operación. La piel puede reaccionar de forma muy sensible a esto. Para que las nalgas no duelan, es aconsejable cuidar especialmente la piel (especialmente en la zona del pañal).
- Medicamentos: En las primeras semanas después de la operación, los niños reciben medicamentos que afectan la consistencia de las heces. Es importante que los niños afectados tomen el medicamento con regularidad.
- Prevención de la formación de cicatrices: después de la operación, es necesario expandir el sitio quirúrgico en el intestino para que el sitio no cicatrice y, por lo tanto, se vuelva a estrechar. Esto se hace con alfileres especiales que estiran el tejido.
- Si se ha creado un ano artificial, se volverá a extraer después de tres o cuatro semanas.
Evitar
Dado que la enfermedad de Hirschsprung es una enfermedad congénita, no existen medidas para prevenir la enfermedad.
Etiquetas: adolescente protección de la piel entrevista