Síndrome de Kallmann
Astrid Leitner estudió medicina veterinaria en Viena. Después de diez años en la práctica veterinaria y el nacimiento de su hija, se pasó, más por casualidad, al periodismo médico. Rápidamente quedó claro que su interés por los temas médicos y su amor por la escritura eran la combinación perfecta para ella. Astrid Leitner vive con una hija, un perro y un gato en Viena y Alta Austria.
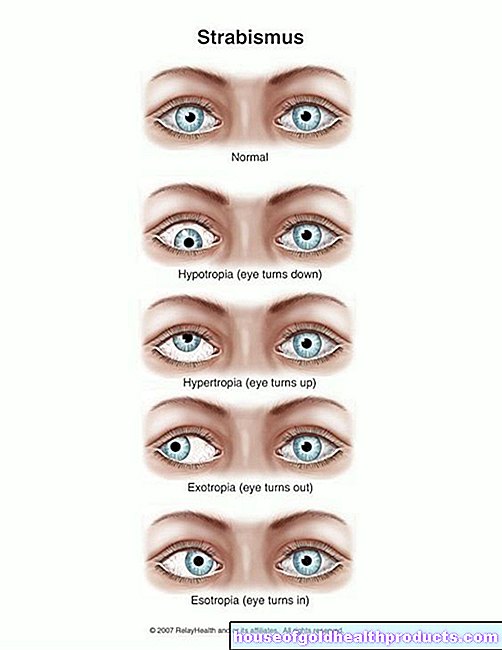
Más sobre los expertos de Todo el contenido de es verificado por periodistas médicos.El síndrome de Kallmann es una enfermedad congénita en la que se producen pocas hormonas sexuales o ninguna. Los afectados no pasan por la pubertad; si no se tratan, tanto los hombres como las mujeres quedan estériles. Además, el sentido del olfato suele verse alterado: los pacientes con SK solo huelen muy poco o nada en absoluto. ¡Lea todo lo que necesita saber sobre el “síndrome olfactogenital” aquí!
Códigos ICD para esta enfermedad: los códigos ICD son códigos reconocidos internacionalmente para diagnósticos médicos. Se pueden encontrar, por ejemplo, en cartas médicas o en certificados de incapacidad laboral. E23

Breve descripción
- ¿Qué es el síndrome de Kallmann? Trastorno congénito del desarrollo que conduce a la falta de hormonas sexuales y, por tanto, a la ausencia de pubertad. Además, la mayoría de los pacientes carecen del sentido del olfato.
- Causas: cambios genéticos congénitos (mutaciones)
- Factores de riesgo: la enfermedad se presenta en familias en alrededor del 30 por ciento de los pacientes.
- Síntomas: Falta de desarrollo de la pubertad (subdesarrollo del pene, testículos y próstata, poco vello púbico, axilar y corporal, falta de crecimiento de la barba, ausencia del primer período menstrual), infertilidad, falta de placer, el sentido del olfato está ausente o grande. consecuencia reducida a largo plazo: osteoporosis
- Diagnóstico: síntomas físicos como falta de desarrollo de características sexuales secundarias, análisis hormonal, prueba genética, ultrasonido, tomografía por resonancia magnética, tomografía computarizada, radiografía
- Tratamiento: terapia de reemplazo con medicamentos hormonales.
- Prevención: no es posible la prevención
¿Qué es el síndrome de Kallmann?
El síndrome de Kallmann (SK, síndrome olfactogenital, síndrome de De-Morsier-Kallmann) es un trastorno congénito del desarrollo del cerebro. Asegura que los afectados no produzcan hormonas sexuales y por tanto no se produzca la madurez sexual. Además, la mayoría de los pacientes con SK sufren de anosmia, lo que significa que no perciben ningún olor.
La causa de la enfermedad es un cambio genético (mutación) que ya afecta el desarrollo embrionario en el útero. En el centro de control hormonal del cerebro (hipotálamo) faltan ciertas células que son responsables del control primordial de la producción de hormonas sexuales en la edad adulta. Además, la modificación del gen significa que el centro olfatorio del cerebro (bulbo olfatorio) no está completamente desarrollado o solo lo está de manera incompleta. Las personas afectadas no huelen nada o solo en un grado muy limitado.
La enfermedad lleva el nombre del psiquiatra alemán Franz Josef Kallmann, quien la describió en 1944 como "hipogonadismo (falta de hormonas sexuales) y anosmia (falta del sentido del olfato)". El término sinónimo “síndrome de De Morsier-Kallmann” también se refiere al neurólogo suizo Georges de Morsier, quien también realizó estudios sobre el síndrome de Kallmann.
Hipogonadismo secundario
El síndrome de Kallmann es una subforma de hipogonadismo secundario. Esto significa una falta de hormonas sexuales, que se desencadena por un mal funcionamiento en el centro de control hormonal en el cerebro (glándula pituitaria). Las gónadas (ovarios, testículos) funcionan normalmente, pero reciben poca o ninguna señal sobre la producción de hormonas sexuales. Si hay anosmia (falta del sentido del olfato) además de la deficiencia hormonal, los médicos hablan del síndrome de Kallmann.
frecuencia
El síndrome de Kallmann es una enfermedad rara; los hombres se ven afectados con más frecuencia que las mujeres: en promedio, alrededor de uno de cada 10.000 hombres y una de cada 50.000 mujeres lo contraen.
Síntomas
Los principales síntomas del síndrome de Kallmann son un sentido del olfato muy reducido o ausente y la ausencia de desarrollo de la pubertad. La gravedad de los síntomas varía de un paciente a otro. Si no se trata, ambos sexos permanecen estériles.
Falta de pubertad: las hormonas sexuales son esenciales para el inicio de la pubertad. Si hay muy pocas hormonas sexuales o ninguna, no se producirá la pubertad y, por tanto, el desarrollo de características sexuales secundarias.
Síntomas en niños:
- Poco o nada de vello púbico, axilar y corporal
- Barba perdida
- No romper su voz
- Micropene y testículos pequeños: en los hombres jóvenes, el pene y los testículos no se desarrollan.
- Testículos no descendidos (a menudo evidentes en la infancia)
- Estatura alta: en las personas sanas, las placas de crecimiento de los huesos largos se cierran tan pronto como se alcanza un determinado nivel de hormonas sexuales. Si faltan las hormonas, los afectados crecen de manera desproporcionada. Son más altos que sus padres y tienen brazos y piernas largos.
Síntomas en niñas:
En las niñas, los síntomas suelen ser menos pronunciados. A menudo, el único síntoma es la ausencia de la primera menstruación (amenorrea primaria), razón por la cual la enfermedad suele reconocerse tardíamente. A diferencia de los niños, el desarrollo físico es en gran parte normal a pesar de la enfermedad. Por ejemplo, los senos suelen desarrollarse con normalidad.
Síntomas en hombres adultos:
Los hombres con síndrome de Kallmann suelen tener una densidad ósea (osteoporosis) y una masa muscular reducidas. A menudo tienen un aspecto externo femenino debido a la distribución de la grasa femenina. Tiene disfunción eréctil y es estéril.
Sentido del olfato disminuido o ausente: los pacientes con SK no perciben los olores (anosmia) o solo perciben los olores débilmente (hiposmia). Este síntoma suele pasar desapercibido porque las personas están acostumbradas a no oler nada desde que nacen. La falta de sentido del olfato puede dar lugar a situaciones peligrosas en casos individuales, por ejemplo, si los afectados no perciben el olor a quemado o el olor a comida en mal estado.
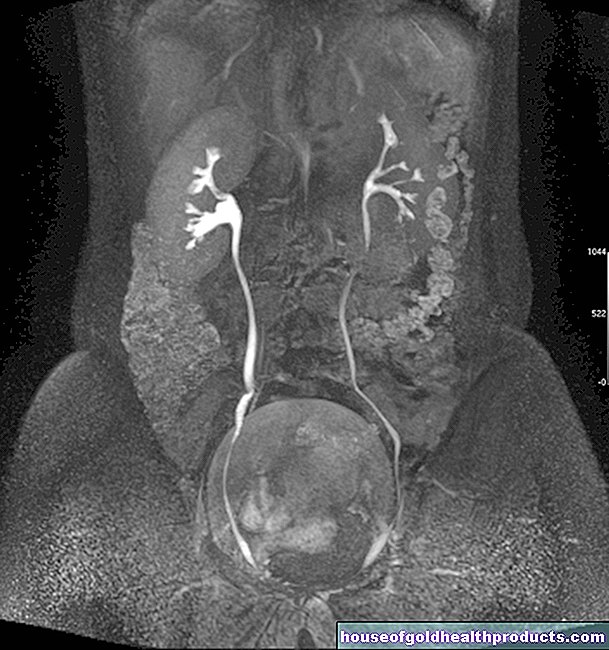
Malformaciones: otras malformaciones físicas ocurren con menos frecuencia en pacientes con SK. Estos son, por ejemplo, trastornos de la audición, labio leporino, mandíbula y paladar hendido o dientes perdidos. Algunas personas nacen con un solo riñón. El desarrollo mental suele ser normal en el SK.
Osteoporosis: las hormonas sexuales como la testosterona y el estrógeno juegan un papel importante en la mineralización ósea. Si faltan las hormonas, los huesos no son tan estables como en las personas sanas; aumenta el riesgo de fracturas óseas.
Causa y factores de riesgo
La causa del síndrome de Kallmann es un cambio genético innato (mutación).Por lo general, surge de forma espontánea, en alrededor del 30 por ciento de los casos se transmite a la descendencia de uno o ambos padres.
El cambio genético ya afecta al desarrollo embrionario en el útero: las células del olfato y las encargadas de controlar las gónadas (ovarios, testículos) surgen de células progenitoras comunes.
En el síndrome de Kallmann, el desarrollo de estas células progenitoras se ve afectado por el cambio genético. Como resultado, las células responsables del control absoluto de la producción de hormonas sexuales y las células olfativas están insuficientemente desarrolladas. Las personas afectadas no desarrollan la pubertad y no perciben olores.
Hormonas sexuales
El hipotálamo (una región del diencéfalo) es el centro de control hormonal del cuerpo. En personas sanas, el hipotálamo libera la hormona GnRH (hormona liberadora de gonadotropinas). Esto, a su vez, estimula la glándula pituitaria para que libere las hormonas LH (hormona luteinizante) y FSH (hormona estimulante del folículo).
La LH y la FSH actúan sobre las gónadas (ovarios, testículos), que finalmente producen las hormonas sexuales: en las mujeres, las hormonas sexuales femeninas estrógeno y progesterona promueven la maduración de los óvulos y desencadenan la ovulación, en los hombres la hormona testosterona provoca la formación de esperma.
Durante la pubertad comienza la producción de hormonas sexuales y con ella la maduración sexual. Si, como en el síndrome de Kallmann, hay poca o ninguna GnRH presente, se producen muy pocas o ninguna hormonas sexuales y no se desarrolla la pubertad. Si no se trata, los afectados no experimentan la madurez sexual y permanecen estériles.
Factores de riesgo
En la mayoría de los casos, el cambio genético se produce de forma espontánea. No está claro cómo ocurre esto. En alrededor del 30 por ciento de todos los casos, la enfermedad ocurre en familias: los afectados han heredado la mutación de uno o ambos padres.
Hasta el momento se han descrito varias mutaciones que provocan el síndrome de Kallmann. Estos incluyen las mutaciones con los nombres KAL1, FGFR1, FGF8, CHD7, SOX10, PROKR2 y PROK2.
Investigación y diagnóstico
El diagnóstico del síndrome de Kallmann se hace tarde en muchos casos, ya que los síntomas a menudo solo se manifiestan en la adolescencia. Esto se aplica especialmente a los niños, en quienes la falta de desarrollo sexual suele ser más claramente visible. En las niñas, los síntomas suelen ser menos pronunciados, por lo que solo la ausencia del primer período menstrual da lugar a una visita al médico. Además, en muchos casos, los afectados no sospechan que haya una enfermedad detrás de los síntomas, sino que creen que tienen un "desarrollo tardío".
En algunos casos, la enfermedad ya es evidente en los bebés: los niños afectados pueden tener testículos no descendidos (criptorquidia) y / o un pene muy pequeño (micropene).
El primer punto de contacto si hay algún signo de síndrome de KS es el pediatra, en el caso de ausencia involuntaria de hijos, el ginecólogo o el urólogo.
El médico realiza los siguientes exámenes:
Antecedentes familiares: si se sospecha el síndrome de Kallmann, el médico pregunta si hay algún caso conocido de SK en la familia.
Examen físico: las características sexuales secundarias poco desarrolladas, como un pene demasiado pequeño para su edad o la ausencia del primer período menstrual, le dan al médico las primeras pistas sobre el síndrome de KS. También presta atención al tamaño del cuerpo, las axilas, el pecho y el vello púbico y evalúa el crecimiento de la barba.
Prueba del olfato: esta prueba es posible a partir de los cinco años de edad. El médico utiliza fragancias puras como la vainillina para comprobar si el paciente puede percibir el olor.
Análisis de sangre: el médico determinará un nivel hormonal modificado con un análisis de sangre. Normalmente, los niveles de GnRH, LH y FSH están disminuidos o en un nivel normal bajo. Los niveles de hormonas sexuales en niños y niñas en la adolescencia son prepúberes.
Examen de ultrasonido de los testículos: el médico usa ultrasonido para examinar los tejidos blandos, como los testículos.
Tomografía computarizada (TC) o tomografía por resonancia magnética (MRT): con estos procedimientos de examen, el médico verifica si el centro olfatorio en el cerebro (bulbo olfatorio) está desarrollado.
Examen de rayos X de la mano: Con la ayuda de un examen de rayos X de la mano, el médico determina si las placas de crecimiento ya están cerradas y, por lo tanto, el cuerpo ha terminado de crecer.
Espermiograma: el médico examina si hay espermatozoides en la eyaculación.
Prueba genética: la prueba genética se utiliza para determinar la mutación exacta que está causando la enfermedad.
¿Cómo se trata el síndrome de Kallmann?
El síndrome de Kallmann es fácilmente tratable. Los afectados reciben un tratamiento sustitutivo con hormonas sexuales. Si la enfermedad se reconoce y se trata antes de la pubertad, los afectados suelen llevar una vida en gran medida sin restricciones. Esto incluye el desarrollo regular de la pubertad y una vida sexual normal.
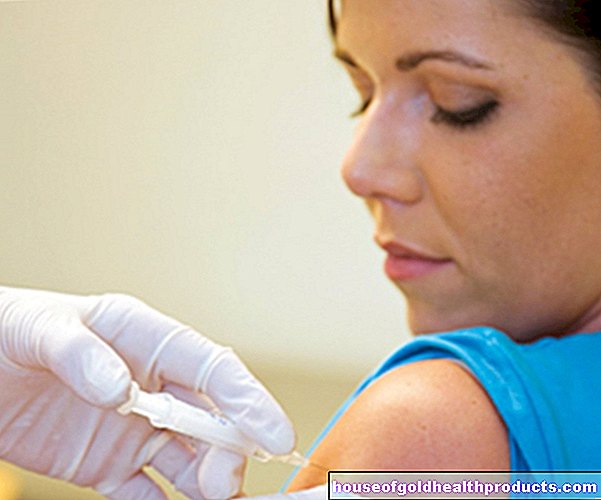
Administración de hormonas sexuales: los hombres reciben testosterona, las mujeres estrógenos y progesterona. Las preparaciones hormonales están disponibles en forma de inyecciones, geles o parches. La terapia hormonal generalmente se continúa de por vida en los hombres y hasta la menopausia en las mujeres.
En el 10 al 20 por ciento de los casos, una deficiencia congénita de GnRH se resuelve después de que finaliza la terapia de reemplazo hormonal. Después de la terapia, los pacientes tienen niveles hormonales normales y experimentan una madurez sexual normal. Por esta razón, los médicos recomiendan suspender la terapia cada uno o dos años para determinar la necesidad de continuarla.
Dar hormonas sexuales si quiere tener hijos: el cuerpo necesita GnRH para que se forme el esperma. Por esta razón, los hombres que desean tener un hijo reciben la hormona GnRH en lugar de testosterona. Se necesitan de 18 a 24 meses para que comience la producción de esperma. En aproximadamente el 80 por ciento de los casos, los hombres son fértiles después. Los hombres con testículos no descendidos tienen un pronóstico ligeramente menos favorable.
Tratamiento de la osteoporosis: los pacientes con densidad ósea reducida reciben calcio y vitamina D. Además, se recomienda el deporte y el ejercicio para fortalecer los huesos.
Anosmia: actualmente no existe una terapia para restaurar el sentido del olfato.
Psicoterapia: para algunos pacientes con SK, la enfermedad es una carga psicológica grave. El psicoterapeuta es el contacto adecuado aquí.
Evolución de la enfermedad y pronóstico.
La forma en que progresa el síndrome de Kallmann varía de una persona a otra. Los síntomas varían de un paciente a otro. Es posible que la enfermedad ya se note en la infancia, se diagnostique en la adolescencia, o que la deficiencia hormonal solo aparezca en la edad adulta.
Si la enfermedad se diagnostica a tiempo, el pronóstico es muy bueno. La maduración sexual se consigue en todos los pacientes con un tratamiento hormonal adecuado. Los pacientes experimentan un desarrollo regular de la pubertad, son fértiles y tienen una esperanza de vida normal. Casi todos los pacientes que desean tener hijos se vuelven fértiles con el tratamiento adecuado.
Si el KS solo se reconoce y se trata después de los 16 años, es posible que ya haya crecido. Este cambio no se puede revertir ni siquiera con medicamentos.
Evitar
Dado que es una enfermedad genética, no es posible la prevención.
Etiquetas: cabello prevención de fumar