hemofilia
y Martina Feichter, editora médica y biólogaDr. medicina Fabian Sinowatz es autónomo en el equipo editorial médico de
Más sobre los expertos deMartina Feichter estudió biología con una asignatura optativa de farmacia en Innsbruck y también se sumergió en el mundo de las plantas medicinales. De ahí no fue lejos para otros temas médicos que aún la cautivan hasta el día de hoy. Se formó como periodista en la Academia Axel Springer en Hamburgo y ha estado trabajando para desde 2007, primero como editora y desde 2012 como escritora independiente.
Más sobre los expertos de Todo el contenido de es verificado por periodistas médicos.
La hemofilia (enfermedad de la sangre) es un trastorno de la coagulación de la sangre que generalmente se hereda. Los enfermos carecen de importantes factores de coagulación sanguínea o presentan defectos. Como resultado, los hemófilos son propensos a sangrar y lesionarse con facilidad. La hemofilia aún no se ha curado. Sin embargo, gracias a las terapias modernas, los hemofílicos pueden llevar una vida en gran parte normal. Lea todo lo que necesita saber sobre la hemofilia aquí.
Códigos ICD para esta enfermedad: los códigos ICD son códigos reconocidos internacionalmente para diagnósticos médicos. Se pueden encontrar, por ejemplo, en cartas médicas o en certificados de incapacidad laboral. D66D67D68
Breve descripción
- ¿Qué es la hemofilia? Un trastorno genético de la coagulación de la sangre (coagulopatía). También se llama hemofilia.
- Formas de hemofilia: la más común es la hemofilia A, seguida de la hemofilia B. Otras formas como la hemofilia C, el síndrome de von Willebrand-Jürgens y la parahemofilia son menos comunes.
- Causa: Deficiencia o defecto de factores de coagulación (proteínas sanguíneas que son necesarias para que la sangre se coagule). En su mayoría se hereda y rara vez se adquiere (causado por una mutación genética espontánea).
- Síntomas: aumento de la tendencia a sangrar, que puede provocar fácilmente hemorragias y hematomas (hematomas). El sangrado también tarda más de lo normal. La gravedad de los síntomas depende de la gravedad de la hemofilia.
- Diagnóstico: medición de varios parámetros sanguíneos (aPTT, valor rápido, tiempo de trombina plasmática, tiempo de sangrado, número de plaquetas sanguíneas), determinación de la actividad de factores de coagulación
- Tratamiento: reemplazo del factor de coagulación faltante (en forma de concentrados de factor); en ciertos casos, otros medicamentos (como desmopresina para la hemofilia A leve)
Hemofilia: descripción
La hemofilia es un trastorno congénito de la coagulación de la sangre. Los afectados (hemofílicos, "hemofílicos") no pueden formar factores de coagulación suficientemente funcionales. Estas son proteínas en la sangre que son necesarias para que la sangre se coagule. Debido a la falta de factores de coagulación, los coágulos de sangre y las heridas no se pueden formar fácilmente. Por lo tanto, los pacientes con hemofilia son propensos a sufrir hemorragias. También tienen heridas que sangran más de lo normal. Esto puede resultar peligroso en determinadas circunstancias.
El término médico para un trastorno de la coagulación de la sangre con una mayor tendencia a sangrar es "diátesis hemorrágica". El nombre general de un trastorno de la coagulación es "coagulopatía".
Hemofilia: frecuencia
La hemofilia ocurre casi exclusivamente en niños y hombres. Es relativamente raro: solo alrededor de dos de cada 10,000 hombres tienen hemofilia. Hay alrededor de 10.000 personas afectadas en Alemania. Alrededor de 3000 a 5000 de ellos padecen una forma grave de la enfermedad.
Hemofilia: formas
Existen diferentes factores de coagulación. Dependiendo de qué factor de coagulación se vea afectado en la hemofilia, los profesionales médicos diferencian entre las diferentes formas de la enfermedad.
Hemofilia A.
En la hemofilia A hay problemas con el factor de coagulación VIII (globulina antihemofílica A): o el cuerpo no puede producirlo en cantidades suficientes o es defectuoso. Aproximadamente el 85 por ciento de todos los hemofílicos padecen hemofilia A. Casi todos son hombres.
Hemofilia B.
En la hemofilia B, falta el factor de coagulación IX (globulina B antihemofílica o factor de Christmas). Aquí, también, los pacientes son en su mayoría hombres. En el pasado, la hemofilia B se presentaba con mayor frecuencia en la familia real inglesa y en la familia zarista rusa. Por eso también se le llama la "enfermedad de los reyes".
Otras formas de hemofilia
Además de las principales formas de hemofilia A y B, existen otros trastornos de la coagulación hereditarios. Uno de ellos es el síndrome de von Willebrand-Jürgens, también llamado síndrome de von Willebrand (vWS). El enfoque aquí está en el factor von Willebrand: este factor de coagulación es demasiado pequeño o defectuoso. Al igual que con la hemofilia A y B, esto conduce a una mayor tendencia a sangrar (diátesis hemorrágica). En Alemania, el síndrome de von Willebrand-Jürgens se encuentra en hasta el uno por ciento de la población. A diferencia de las dos formas principales de hemofilia, este trastorno de la coagulación afecta por igual a hombres y mujeres.
En casos raros, también se producen otros trastornos de la coagulación, que se basan en la falta de factores de coagulación. También tienen una mayor tendencia a sangrar (diátesis hemorrágica). Éstos incluyen:
- Hemofilia C: deficiencia del factor funcional XI
- Parahemofilia: falta de funcionamiento del factor V
- Hipoproconvertinemia: deficiencia de factor VII funcional
- Deficiencia del factor Stuart Prower: falta de factor de trabajo X
- Síndrome de Hageman: deficiencia del factor XII funcional
- Deficiencia de fibrinasa: deficiencia del factor XIII funcional
Hemofilia: síntomas
Los síntomas de la hemofilia A (deficiencia de factor VIII) y la hemofilia B (deficiencia de factor IX) son los mismos, incluso si se ven afectados diferentes factores de coagulación en cada caso. En general, se puede decir: cuantos menos factores de coagulación funcionales haya, más pronunciado será el cuadro clínico.
Hemofilia: grados de gravedad
La gravedad de la hemofilia depende de cuánto se reduzca la actividad de los factores de coagulación en comparación con su función en personas sanas. Si la actividad del factor se reduce solo ligeramente, los afectados generalmente no presentan ningún síntoma. Por otro lado, en los hemófilos sintomáticos, la actividad del factor es más reducida, lo que provoca signos más pronunciados. Hay tres grados de gravedad de la hemofilia (A y B):
Hemofilia leve:La actividad del factor es del 6 al 45 por ciento de la actividad normal de una persona sana; alrededor del 20 por ciento de todas las personas con hemofilia tienen esta gravedad. Los afectados a menudo no notan mucho de su enfermedad. Es por eso que la hemofilia solo se descubre en muchas personas en la adolescencia o la edad adulta, cuando las operaciones o las lesiones graves sangran más de lo esperado.
Hemofilia moderada: La actividad de los factores es del 1 al 5 por ciento de la actividad normal; aproximadamente el 20 por ciento de todos los hemófilos también se ven afectados. Los síntomas generalmente se vuelven obvios en los primeros años de vida con sangrado inusualmente prolongado y hematomas frecuentes. Al igual que con la hemofilia leve, el sangrado suele ser el resultado de lesiones o cirugía. El sangrado espontáneo es raro.
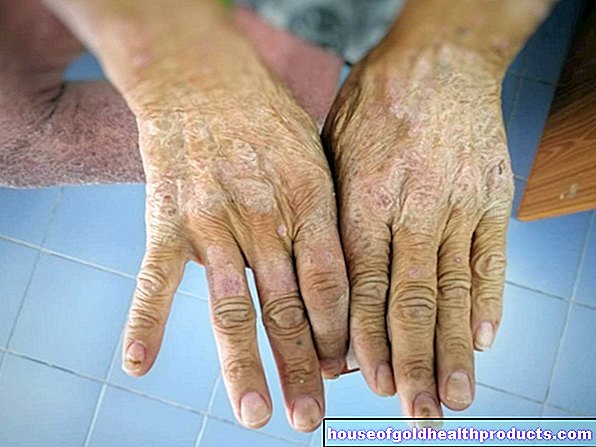
Hemofilia severa: La actividad de los factores es menos del 1 por ciento de la actividad normal. Esto se aplica a aproximadamente el 55 por ciento de todos los hemofílicos. La hemofilia generalmente ya se nota al nacer: la extirpación del cordón umbilical desencadena un sangrado masivo. Las hemorragias nasales abundantes no son infrecuentes en la infancia. Además, incluso las lesiones o los golpes más pequeños pueden causar un sangrado profuso debajo de la piel (hematomas). El sangrado interno también es más común, por ejemplo, sangrado doloroso en las articulaciones grandes (como rodillas, codos). Por lo general, muchas hemorragias no tienen una causa aparente (hemorragia espontánea).
Hemofilia: riesgos y peligros
El sangrado en las articulaciones (hemartrosis) a menudo ocurre repetidamente, especialmente en la hemofilia severa. Como resultado, la articulación afectada puede deformarse, desgastarse prematuramente (osteoartritis) y endurecerse gradualmente. Las personas con hemartosis avanzada apenas pueden mover los brazos y las piernas y dependen de una silla de ruedas.
Otro peligro de la hemofilia es la hemorragia en los músculos: puede dañar el tejido muscular y provocar debilidad muscular.
En principio, la hemorragia interna en la hemofilia puede ocurrir en cualquier área de órganos. El sangrado en el cerebro es raro pero peligroso: por ejemplo, puede afectar la capacidad de pensar y la concentración. ¡La hemorragia cerebral severa puede incluso ser fatal! El sangrado en el abdomen también puede poner en peligro la vida en determinadas circunstancias. El sangrado abundante en la orofaringe puede afectar las vías respiratorias.
Además de las tendencias hemorrágicas (diátesis hemorrágica), la hemofilia de cualquier grado de gravedad también puede provocar trastornos en la cicatrización de heridas.
Síndrome de Von Willebrand-Juergens: síntomas
Las personas con síndrome de von Willebrand-Jürgens también tienen una mayor tendencia a sangrar. La mayoría de las veces, hay un ligero sangrado debajo de la piel (hematomas), encías sangrantes o sangrado prolongado después de la cirugía, por ejemplo, después de una extracción dental. Pero también hay pacientes que sufren hemorragias abundantes.
Hemofilia: tratamiento
La terapia de la hemofilia depende del tipo y la gravedad de la hemofilia. El médico tratante creará un plan de terapia adecuado para cada paciente. Además del tratamiento farmacológico, también puede recomendar medidas generales. Por ejemplo, puede ser recomendable, especialmente en el caso de hemofilia severa, tener cuidado físico y evitar ciertos deportes (lesivos).
Hemofilia A y B
Hoy en día, los denominados concentrados de factor están disponibles para el tratamiento de la hemofilia. Estos son concentrados de factores de coagulación sanguínea VIII (para la hemofilia A) o IX (para la hemofilia B) obtenidos del plasma sanguíneo o modificados genéticamente. Deben inyectarse en una vena (por vía intravenosa). Muchos pacientes aprenden a inyectarse el concentrado de factor ellos mismos. Esto les da mucha independencia para hacer frente a la enfermedad.
En las décadas de 1960 y 1970, muchas personas en Alemania contrajeron hepatitis y / o VIH a partir de preparaciones de factor contaminado. Esto prácticamente ya no puede suceder hoy: el plasma sanguíneo está estrictamente controlado y pretratado antes de su administración. Y con los concentrados de factor modificados genéticamente, generalmente no hay riesgo de infección.
En el caso de hemofilia leve y moderada, la administración de concentrado de factor solo es necesaria si es necesario (tratamiento a demanda): El anticoagulante se administra, por ejemplo, en caso de sangrado más abundante o antes de una operación planificada. Las lesiones menores, como las abrasiones, no necesitan tratarse con concentrado de factor. Por lo general, el sangrado se puede detener aplicando una ligera presión en el área que sangra.
Las personas con hemofilia grave, por otro lado, deben inyectarse concentrado de factor regularmente (tratamiento a largo plazo). El factor VIII en la hemofilia A se administra dos o tres veces por semana. El factor IX en la hemofilia B generalmente solo necesita inyectarse una o dos veces por semana debido a su mayor tiempo de retención en la sangre.
Cirugías y lesiones agudas
Antes de las operaciones (incluso si se planea la extracción de un diente), se debe realizar una terapia preparatoria para todos los pacientes con hemofilia. Por lo general, se administra un concentrado de factor para este propósito. Ésta es la única forma de prevenir complicaciones graves por una pérdida excesiva de sangre durante la cirugía.
En el caso de la hemofilia A leve, en lugar de concentrados de factor, se puede administrar un medicamento que estabilice la coagulación de la sangre antes de una operación planificada. Esto incluye desmopresina, por ejemplo. Esta es una proteína producida artificialmente. Estimula la liberación de factor VIII almacenado de los vasos sanguíneos. El medicamento solo se puede usar durante unos días, de lo contrario, las tiendas pronto estarán vacías.
Consejo: antes de un procedimiento planificado, las personas con hemofilia deben consultar con su hematólogo o centro de hemofilia qué terapia preventiva es aconsejable en su caso.
En el caso de lesiones agudas en una emergencia, además de las medidas de hemostasia local (por ejemplo, vendajes de presión), también es necesaria la administración de concentrado de factor.
Concentrado de factor: complicaciones
Algunas personas forman anticuerpos (inhibidores) contra los factores de coagulación en el concentrado de factor. Este llamado inhibidor de la hemofilia es significativamente más común en personas con hemofilia A que en personas con hemofilia B. Los inhibidores inactivan el factor de coagulación agregado. Entonces, la terapia no es tan eficaz como se desea. La hemofilia B también amenaza con reacciones alérgicas graves y otras complicaciones.
La cantidad de inhibidores en sangre se administra en la denominada unidad Bethesda (BE). Cuanto mayor sea el valor de BE, más inhibidores hay en la sangre del paciente.
En la hemofilia A, una ligera acumulación de inhibidores a menudo se puede compensar aumentando la dosis de concentrado de factor. Si los inhibidores se forman en grandes cantidades, se recomienda la terapia de tolerancia inmunitaria: el paciente recibe dosis muy altas del factor de coagulación faltante en un régimen de terapia compleja. El sistema inmunológico debe acostumbrarse gradualmente a su presencia y detener la formación de inhibidores.
El raro inhibidor de la hemofilia en la hemofilia B se trata de manera diferente. Por ejemplo, los afectados reciben medicamentos que afectan el sistema inmunológico (inmunomoduladores).
Analgésico
La hemofilia grave puede causar un dolor intenso a los afectados. Por ejemplo, sangrar en las articulaciones puede ser muy doloroso. Entonces ayudan los analgésicos como el ibuprofeno. Por el contrario, el analgésico ácido acetilsalicílico (AAS) no es adecuado para la hemofilia: aumenta la tendencia a sangrar aún más (efecto secundario del AAS).
Síndrome de von Willebrand-Juergens (VWS)
En el síndrome de von Willebrand-Jürgens se distingue entre diferentes tipos, que se tratan de forma diferente: en el denominado tipo 1, el principio activo desmopresina se administra cuando es necesario (antes de una operación o en caso de hemorragia aguda). Estimula la liberación de factores de coagulación almacenados.
La desmopresina también se usa en el tipo 2; sin embargo, la droga no siempre funciona aquí. Luego, el médico prescribe un concentrado de factor (con factor von Willebrand) en su lugar.
Los pacientes con VWS tipo 3 siempre se tratan con un concentrado de factor.
Hemofilia: causas y factores de riesgo
La hemofilia es un trastorno genético congénito que generalmente se hereda. Ocurre más raramente de forma espontánea (como resultado de un cambio genético espontáneo = mutación espontánea).
En hemofílicos, la información genética requerida para producir un factor de coagulación funcional es incorrecta: en hemofilia A, es factor de coagulación VIII, en hemofilia B, factor IX. El resultado de un plan de construcción incorrecto es que el factor de coagulación relevante no se puede producir en una cantidad suficientemente funcional. Esto interrumpe la coagulación de la sangre: las heridas no se cierran tan rápido, por lo que el sangrado lleva un tiempo inusualmente largo. Algunos pacientes también presentan sangrado espontáneo (sin causa aparente).
Hemofilia A y B: herencia
Los planos (genes) de todas las partes del cuerpo se encuentran en los cromosomas. En el núcleo de cada célula del cuerpo hay 46 cromosomas, incluidos dos cromosomas sexuales, que, entre otras cosas, determinan el género. Las mujeres tienen dos cromosomas sexuales X (XX): un cromosoma X cada uno fue heredado de la madre y otro del padre. Los hombres, por otro lado, tienen un cromosoma Y heredado del padre y un cromosoma X heredado de la madre (XY).
Los genes de los factores de coagulación se encuentran en el cromosoma X. En las mujeres que tienen dos cromosomas X, si uno de ellos contiene un modelo defectuoso de un factor de coagulación, esto generalmente puede compensarse con el otro cromosoma X. Por lo tanto, están prácticamente libres de síntomas durante toda su vida.
Si una mujer así tiene un hijo, le transmite uno de los dos cromosomas X a la descendencia. La probabilidad de que esta sea la copia con el plano defectuoso es del 50 por ciento. Al transmitir el defecto genético, la madre se convierte en portadora (portadora) de la hemofilia. Si hay un hijo, nacerá hemofílico. Una hija, por otro lado, generalmente se convierte a su vez en una portadora potencial.
En casos raros, el factor de coagulación relevante no se forma suficientemente incluso en conductores femeninos. Las heridas producidas por lesiones u operaciones pueden sangrar durante mucho tiempo. Es aún más raro que una niña herede un cromosoma X defectuoso de ambos padres. Esto se debe principalmente a la enfermedad hereditaria síndrome de Turner. Las niñas afectadas muestran el cuadro completo de la hemofilia.
Síndrome de von Willebrand-Juergens
En el síndrome de von Willebrand-Jürgens, las instrucciones para el factor von Willebrand (vWF) muestran una mutación: el factor de coagulación es demasiado pequeño o está defectuoso. La mutación genética puede ocurrir tanto en hombres como en mujeres.
Hemofilia: exámenes y diagnóstico
Si alguien tiene hemorragias espontáneas frecuentes (como hemorragias nasales) o se forman moretones con mucha facilidad, esto podría ser un indicio de hemofilia. Esta sospecha es particularmente obvia si la familia ya ha conocido casos de hemofilia. Los afectados deben hacer que un médico aclare el aumento de la tendencia al sangrado. El primer punto de contacto si sospecha de hemofilia es su médico de cabecera:
El médico primero tomará el historial médico del paciente (anamnesis) en conversación con el paciente: tiene los síntomas descritos en detalle, pregunta sobre cualquier enfermedad subyacente y si hay algún caso conocido de hemofilia en la familia.
Las pruebas de laboratorio son particularmente importantes para aclarar una posible hemofilia. El médico toma una muestra de sangre del paciente para examinarla en el laboratorio en busca de varios parámetros: En la hemofilia, el llamado aPTT (tiempo de tromboplastina parcial activada) es más largo que en personas sanas. Por el contrario, el denominado valor Quick (tiempo de tromboplastina, TPZ) y el tiempo de trombina plasmática (PTZ) son generalmente normales; sólo se prolongan en casos de hemofilia grave. La cantidad de plaquetas en sangre (trombocitos) y el llamado tiempo de sangrado también son normales en la hemofilia A y B. En el síndrome de von Willebrand-Jürgens, por otro lado, el tiempo de sangrado se prolonga. El tiempo de sangrado es la cantidad de tiempo que tarda el sangrado en detenerse.
Para poder determinar de forma inequívoca la hemofilia A o B, se debe analizar la actividad de los factores de coagulación (VIII, IX). Un especialista en hematología o un centro médico especializado (centro de hemofilia) se encarga de ello.
Hemofilia: prueba en recién nacidos y bebés por nacer
Si una familia ya ha desarrollado hemofilia, la coagulación de los recién nacidos varones generalmente se controla inmediatamente después del nacimiento. De esta forma, la hemofilia se puede detectar en una etapa temprana. También puede buscar hemofilia durante el embarazo.
Si una mujer sospecha que tiene una predisposición genética a la hemofilia y, por tanto, es una portadora potencial, esto puede aclararse con una prueba genética.
Hemofilia: curso de la enfermedad y pronóstico
La hemofilia aún no se ha curado. Los pacientes tienen que lidiar con la falta de factores de coagulación a lo largo de su vida. Sin embargo, con la ayuda de concentrados de factor, por lo general pueden llevar una vida mayormente normal.
Si no se trata, la hemofilia moderada y grave a menudo conduce a complicaciones graves. Por ejemplo, una hemorragia en los músculos puede causar daño muscular. La hemorragia en las articulaciones puede provocar osteoartritis y rigidez articular. Estas complicaciones pueden evitarse si la hemofilia se detecta a tiempo y se trata de manera constante.
Etiquetas: Diagnóstico prevención estrés