Fenilcetonuria
Marian Grosser estudió medicina humana en Munich. Además, el médico, que estaba interesado en muchas cosas, se atrevió a hacer algunos desvíos apasionantes: estudiar filosofía e historia del arte, trabajar en la radio y, finalmente, también para un Netdoctor.
Más sobre los expertos de Todo el contenido de es verificado por periodistas médicos.La fenilcetonuria (PKU) es una enfermedad congénita y hereditaria del metabolismo de las proteínas. Previene la degradación del aminoácido fenilalanina. Esto se acumula en el cuerpo y altera el desarrollo del cerebro del niño. Si no se trata, la fenilcetonuria conduce a discapacidades intelectuales graves. Sin embargo, con una terapia oportuna, los pacientes pueden llevar una vida normal. ¡Descubra todo sobre la fenilcetonuria aquí!
Códigos ICD para esta enfermedad: los códigos ICD son códigos reconocidos internacionalmente para diagnósticos médicos. Se pueden encontrar, por ejemplo, en cartas médicas o en certificados de incapacidad laboral. E70
Fenilcetonuria: descripción
La fenilcetonuria (PKU) es una enfermedad metabólica hereditaria que existe desde el nacimiento e interfiere con la degradación del aminoácido esencial fenilalanina. Los aminoácidos son los componentes básicos de las proteínas y, por tanto, los componentes metabólicos vitales. Algunos de ellos solo pueden ingresar al cuerpo a través de los alimentos, el organismo no puede producirlos por sí mismo. Estos aminoácidos se denominan esenciales.
¿Qué sucede con la fenilcetonuria?
Normalmente, los aminoácidos están sujetos a un equilibrio entre la absorción / acumulación y la degradación, de modo que siempre haya tanto disponible como necesite el cuerpo. En el caso de varios aminoácidos, una deficiencia o exceso puede causar un daño considerable y causar varios síntomas.
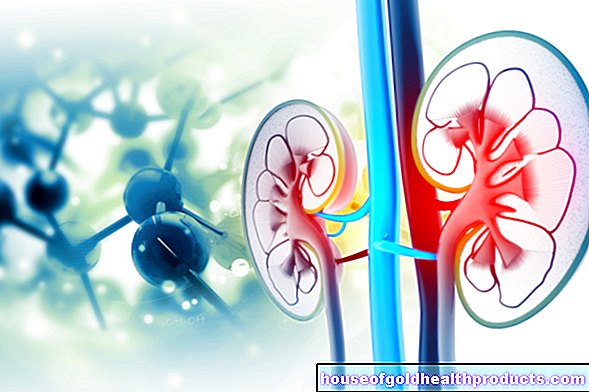
En la denominada PKU clásica, el efecto de la fenilalanina hidroxilasa (PAH) es limitado o incluso completamente ausente. Debido a la deficiencia de PAH, la fenilalanina se acumula cada vez más en el cuerpo. Una concentración demasiado alta de fenilalanina interrumpe considerablemente el desarrollo del cerebro y conduce a discapacidades intelectuales en pacientes jóvenes en una etapa temprana.
Debido a que la descomposición normal de la fenilalanina no es posible con la enfermedad, se forman otros productos de descomposición, las llamadas fenilcetonas. Se excretan en la orina y son responsables del nombre de la enfermedad.
Fenilcetonuria atípica
Incluso con formas atípicas de fenilcetonuria, se altera la degradación de la fenilalanina. Sin embargo, la causa no es un defecto en la HAP. En cambio, la función de una coenzima, tetrahidrobiopterina (BH4), está restringida. Está indirectamente involucrado en la descomposición de la fenilalanina porque el PAH necesita BH4 para convertir la fenilalanina en tirosina.
Dado que la BH4 también es importante para la producción de las sustancias mensajeras dopamina y serotonina, la fenilcetonuria atípica con deficiencia de BH4 suele ser más complicada que la forma clásica.
¿A quién afecta la fenilcetonuria?
La PKU es una de las enfermedades metabólicas congénitas más comunes. Se estima que alrededor de uno de cada 7.000 recién nacidos en todo el mundo lo desarrollará, sin diferencias entre niñas y niños. Dado que es una enfermedad hereditaria, a menudo se ven afectados varios miembros de una familia.
Fenicetonuria: síntomas
Al principio, los bebés con fenilcetonuria no muestran ningún síntoma de la enfermedad. Los primeros problemas no ocurren hasta el cuarto al sexto mes de vida, si la enfermedad aún no ha sido reconocida y tratada. Sobre todo, la maduración cerebral alterada causa complicaciones masivas con el tiempo. Los síntomas de la PKU no tratada incluyen:
- un fuerte déficit mental. El daño cerebral progresa a lo largo de la pubertad y luego se estanca. Los niños afectados suelen tener una discapacidad mental grave.
- Ataques (convulsiones) (epilepsia). Debido al daño, las células nerviosas del cerebro son particularmente sensibles y sobreexcitables. El resultado son ataques epilépticos frecuentes.
- discapacidades motoras. No solo las células cerebrales, sino también los músculos del paciente pueden estar sobreexcitados. Es por eso que a menudo se vuelve tenso (espasticidad), lo que conduce a varios trastornos del movimiento.
- Trastornos de la conducta. Algunos niños con fenilcetonuria son hiperactivos e inusualmente agresivos, y los arrebatos de ira también son más comunes.
- una cabeza pequeña (microcefalia). Debido a que el cerebro del paciente no se desarrolla adecuadamente, el crecimiento de la cabeza también se retrasa. La pequeña circunferencia de la cabeza en comparación con sus compañeros es particularmente notable en los niños mayores.
- un olor perceptible. La PKU produce ciertos productos de degradación de la fenilalanina que huelen de manera similar a los excrementos de ratón. Estas sustancias se excretan principalmente en la orina, pero también en parte a través de la piel.
- cambios en la piel similares a los del eccema
Debido a que la producción del pigmento melanina también se altera en la fenilcetonuria, muchos pacientes tienen la piel muy clara, sensible al sol y el cabello rubio blanquecino. El iris de los ojos también es de azul claro a transparente y permite que el fondo de ojo rojizo brille a través.
Los síntomas de la PKU varían en gravedad de una persona a otra. La razón principal de esto es que la actividad de la fenilalanina hidroxilasa (PAH) está restringida de manera diferente en cada paciente. Algunos todavía tienen cierta actividad residual, por lo que se acumula menos fenilalanina en el organismo. Otros no muestran ninguna actividad enzimática: la enfermedad progresa correspondientemente más rápido y con mayor gravedad.
Fenilcetonuria: causas y factores de riesgo
La fenilcetonuria es una enfermedad hereditaria. Actualmente se conocen numerosas mutaciones genéticas que conducen a un defecto en la HAP. El tipo de mutación determina hasta qué punto se restringe la degradación de la fenilalanina.
La PKU se hereda de forma recesiva, lo que significa que una persona puede ser portadora de un gen alterado sin desarrollar la enfermedad. Asimismo, las personas con fenilcetonuria pueden engendrar hijos sanos.
Solo si ambos padres tienen mutaciones en su composición genética, existe una cierta probabilidad de que su descendencia desarrolle fenilcetonuria. Si los padres no solo son portadores de los genes, sino que también tienen PKU, todos los niños juntos también la desarrollarán.
Fenilcetonuria: exámenes y diagnóstico
Dado que las graves consecuencias de la fenilcetonuria se pueden prevenir iniciando el tratamiento a tiempo, es particularmente importante descubrir la enfermedad lo antes posible. En Alemania, los niños son examinados para detectar diversas enfermedades congénitas, incluida la PKU, como parte de un examen general (cribado neonatal) al tercer día después del nacimiento.
Espectrometría de masas en tándem
Muchos trastornos metabólicos congénitos ahora se diagnostican con la ayuda de la llamada espectrometría de masas en tándem. Permite examinar la sangre del recién nacido de forma rápida y sencilla. Además de la fenilcetonuria, los médicos pueden detectar más de 20 enfermedades más en unos pocos minutos.
Prueba de Guthrie
La prueba de Guthrie, que lleva el nombre de su inventor, también permite diagnosticar la PKU. Para hacer esto, se extrae una pequeña cantidad de sangre del talón del niño y se aplica a un trozo de papel de filtro. En el laboratorio, puede determinar si aumenta la concentración de fenilalanina.
La prueba de Guthrie se introdujo en la década de 1960 y ha sido durante mucho tiempo el método estándar para diagnosticar la fenilcetonuria. Sin embargo, tiene desventajas en comparación con la espectrometría de masas en tándem. La prueba de Guthrie solo arroja un resultado después de cinco días, que también es propenso a errores. Por ejemplo, factores como la dieta del niño o la posible terapia con antibióticos falsean los resultados. En algunos países, la prueba de Guthrie todavía se usa, en Alemania, por lo general, ya no se usa.
Otras pruebas
Si la prueba de detección del recién nacido revela una sospecha de fenilcetonuria, se realiza un examen adicional para confirmarlo. Esto también se puede utilizar para determinar la concentración exacta de fenilalanina en la sangre.
Finalmente, es importante distinguir si se trata de una PKU típica (clásica) o atípica. También se encuentran disponibles pruebas especiales para esto, como la prueba de esfuerzo con tetrahidrobiopterina. La distinción es importante porque la fenilcetonuria atípica se trata de manera diferente a la forma clásica.
Examen de líquido amniótico
La fenilcetonuria ya se puede diagnosticar durante el embarazo (diagnóstico prenatal). Para hacer esto, se extrae una pequeña cantidad de líquido amniótico del saco amniótico de la madre y se examinan las células del feto que contiene. De esta manera se puede identificar cualquier defecto genético que cause PKU.
Sin embargo, con las pruebas de detección del recién nacido, la fenilcetonuria generalmente se descubre lo suficientemente temprano como para ser tratada. Debido a que una prueba de líquido amniótico siempre se asocia con un cierto riesgo, su uso para diagnosticar la PKU generalmente no es útil.
Fenilcetonuria: tratamiento
Solo hay una forma de contrarrestar el exceso de fenilalanina en la PKU: los niños afectados deben seguir una dieta especial para ingerir la menor cantidad posible de fenilalanina con sus alimentos. También necesitan ciertos complementos alimenticios para reemplazar las sustancias que se formarían a partir de la fenilalanina en niños sanos.
La terapia debe comenzar antes de que aparezcan los primeros síntomas de un trastorno del desarrollo, es decir, dentro de los dos primeros meses de vida. El daño cerebral que ya ha ocurrido no se puede revertir.
Terapia nutricional con la dieta PKU
Todas las proteínas naturales consisten en aproximadamente un cinco por ciento del aminoácido fenilalanina. La mayoría de los alimentos contienen mucho más de lo que el cuerpo necesita. Esto no es un problema para una persona sana porque descompone y excreta las cantidades excesivas de fenilalanina.
Los pacientes con fenilcetonuria, por otro lado, tienen que prescindir en su mayor parte de proteínas naturales. Los productos especiales fabricados industrialmente reemplazan los componentes alimentarios que faltan. Por un lado, se quiere evitar un exceso de fenilalanina, por otro lado, se debe suministrar al paciente sustancias que de otro modo faltarían, como la tirosina, que se forma a partir de la fenilalanina.
Sin embargo, el objetivo de la dieta para la PKU no es detener por completo la absorción de fenilalanina. Porque el organismo necesita cierta cantidad del aminoácido para procesos metabólicos importantes. Lograr la concentración adecuada representa un gran desafío para médicos y pacientes y requiere mucha disciplina.
Por lo tanto, la dieta debe seguirse durante toda la vida, pero de forma especialmente estricta hasta los seis años. Porque el cerebro se desarrolla con mucha fuerza hasta esta edad y, por lo tanto, es particularmente susceptible a sufrir daños. En la edad adulta, las altas concentraciones de fenilalanina no causan daño cerebral como lo hacen en los niños. Sin embargo, pueden ser el desencadenante de otras dolencias neurológicas como la falta de concentración o reacciones lentas.
El tratamiento dietético para la fenilcetonuria debe comenzar en un centro especializado en enfermedades metabólicas. Porque no es posible en todas las clínicas instruir a los padres sobre la dieta. Esto se hace mejor con la ayuda de un asesoramiento dietético, que muestra cómo seguir la dieta y controlar regularmente los niveles de fenilalanina en sangre.
Tratamiento de la fenilcetonuria atípica
En las formas atípicas de PKU, la coenzima BH4 faltante y ciertas sustancias mensajeras como la dopamina y la serotonina se reemplazan artificialmente. En algunos casos, el paciente también debe seguir una dieta baja en fenilalanina.
Fenilcetonuria durante el embarazo
Si está embarazada y tiene fenilcetonuria, debe considerar lo siguiente:
- ¡Siga su dieta de manera particularmente estricta!
- Controle sus valores sanguíneos de fenilalanina a intervalos frecuentes. Porque las concentraciones de fenilalanina en el feto son aproximadamente el doble que en la mujer embarazada. Además del cerebro, también pueden dañar el corazón y los ojos del feto. Las malformaciones del esqueleto del niño también pueden deberse a niveles altos de fenilalanina durante el embarazo.
- Para descartar daño cerebral al niño al comienzo del embarazo, las mujeres con fenilcetonuria deben planificar el embarazo con cuidado y tomar precauciones desde el principio.
- En mujeres embarazadas con fenilcetonuria, una dieta que no se adhiera estrictamente puede provocar un aborto espontáneo (aborto espontáneo).
- En cualquier caso, las mujeres embarazadas con fenilcetonuria deben buscar el consejo y la orientación de un médico.
Fenilcetonuria: curso de la enfermedad y pronóstico
Si la fenilcetonuria se diagnostica temprano, si es posible en el recién nacido, y si se sigue la dieta especial para la fenilcetonuria, el pronóstico suele ser bueno. Los niños se desarrollan espiritualmente y tienen una esperanza de vida media.
Sin embargo, si no se trata, el daño cerebral conduce a graves trastornos del desarrollo mental que no se pueden corregir más adelante. Los afectados tienen una esperanza de vida normal, pero su coeficiente de inteligencia casi siempre está muy por debajo de la norma.
La rara forma atípica de fenilcetonuria, en la que hay deficiencia de BH4, es un caso especial. Esta variante de fenilcetonuria puede conducir a un daño neurológico progresivo con calambres severos a pesar de la dieta.
Etiquetas: Sistemas de órganos plantas venenosas de seta venenosa cuidado dental